27Al Chemical Shifts and Quadrupolar Coupling Constants
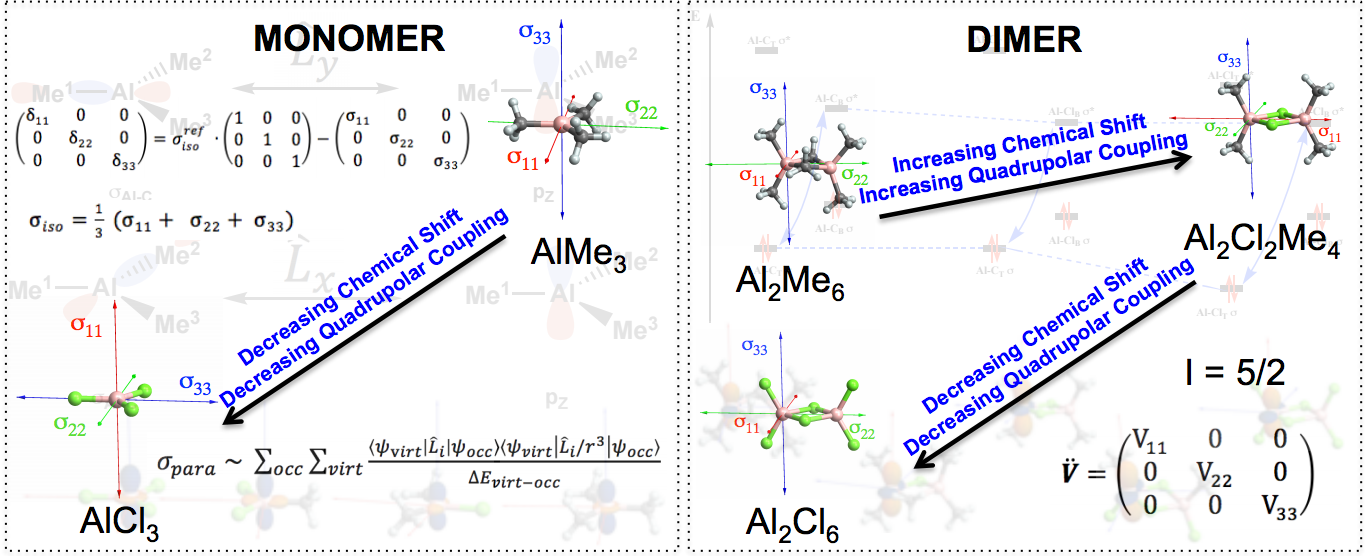
Understanding Trends in 27Al Chemical Shifts and Quadrupolar Coupling Constants in Chloroalkyl Aluminum [AlClx(Me)3‐x]n=1 or 2 Compounds
The usage of alkyl aluminum compounds and related structures as co-catalyst finds a broad range of application in homogeneous and heterogeneous catalysis. While understanding the nature of the aluminum species in solution or in solids can be a challenge, 27Al solid state NMR is a powerful tool to understand the structure of Al species, but their assignment remains mostly empirical, typically by comparing chemical shifts with known compounds. In this work, the observed trends in 27Al NMR parameters - chemical shift and quadrupolar coupling constant - of chloroalkyl aluminum compounds, a prototypical class of important Lewis activators, are traced back to their frontier orbitals and electron polarization through a natural localized molecular orbital analysis. This study thus provides guidelines to understand the nature of chemical shift and thereby assignment of possible structure.
Access the paper published in external pageHelvetica.
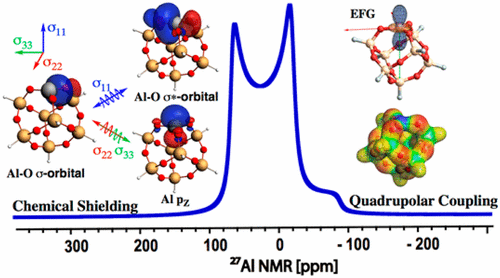
Role of Coordination Number, Geometry, and Local Disorder on 27Al NMR Chemical Shifts and Quadrupolar Coupling Constants: Case Study with Aluminosilicates
27Al-Solid-state NMR is a powerful tool for elucidating local geometries at Al sites in molecular and solid-state systems because they are typically associated with specific NMR signatures, namely isotropic chemical shift (δiso) and quadrupolar coupling constant (CQ). Assignment is however mostly empirical, hence obtaining a detailed understanding of the origins of the NMR parameters would be a valuable step towards structure–property/reactivity relationship. Here, we investigate the origin of the 27Al-NMR signatures in aluminosilicates using DFT calculations on cluster models complemented by Natural Chemical Shift (NCS) analysis. In particular, NCS analysis shows that the chemical shift of Al is mostly associated with the coupling Al-O σ- and σ*–orbitals for σ11 leading to deshielding as the coordination number of Al decreases, allowing the distinction between tri-, tetra-, penta- and hexa-coordinated sites. In contrast, the CQ can take a broad range of values (ranging between 8.0 and 23.6 MHz), independently of the coordination number because it is greatly affected by slight variation of the bond distance of siloxane bonds coordinated to aluminum, which perturbs the electrostatic interaction with aluminum and thereby the CQ.
Access the paper published in external pageThe Journal of Physical Chemistry C.